
Innovative Computational Design of Sustainable Milk-Clotting Peptides for Enhanced Cheese Production
Applied Food Biotechnology,
Vol. 12 No. 1 (2025),
4 January 2025
,
Page 1-30 (e19)
https://doi.org/10.22037/afb.v12i1.48544
Abstract
Background and Objective: Identifying a milk-clotting enzyme (MCE) with high κ-casein specificity and heat sensitivity remains a challenge in cheese production. Current microbial, plant, and recombinant MCEs often exhibit low clotting activity, poor κ-casein specificity, and high thermostability, compromising cheese quality and increasing production costs. In this study, to address this, we developed a computational pipeline combining structural analysis, machine learning, and molecular dynamics simulation to design approximately 160,000 peptides from the Rhizomucor miehei protease–Pepstatin A complex (PDB ID: 2RMP).
Material and Methods: Single-site mutagenesis, ML-driven affinity re-prediction, and physicochemical filtering yielded 84 peptides. Their specificity as aspartic proteases were validated via predicted Pepstatin A binding and further screened for cross-reactivity with αs1-, αs2-, and β-caseins.
Results and Conclusion: Two candidates, Pep1 and Pep2, demonstrated superior κ-casein binding affinities (ΔG = -50.20 and -39.07 kcal/mol at 40 °C, respectively), lower melting indices (-4.05 and -3.09), and significantly enhanced specificity scores (-10.85) compared to Rhizomucor miehei protease (ΔG = -33.9 kcal/mol at 45 °C; melting index = 0.17; specificity score = 0.85). These peptides represent promising vegan- and halal-friendly alternatives to chymosins, pending experimental validation.
Keywords: Milk-clotting enzymes, Computational peptide design, κ-casein specificity, Thermolabile peptides, Sustainable cheese production, Molecular dynamics simulations, Machine learning, Vegan cheese production, Halal cheese production, Aspartic protease.
. Introduction
Identifying chymosin substitutes with comparable enzymatic characteristics particularly high specificity for κ-casein (reflected in a high milk-clotting activity to proteolytic activity ratio, MCA/PA) and optimal heat sensitivity remains a significant challenge in cheesemaking [1-3]. Chymosin, traditionally derived from the stomachs of calves, has long been the gold standard for milk clotting due to its high specificity for κ-casein and its ability to produce high-quality cheese [4]. However, economic, ethical, dietary, and religious concerns, coupled with the rising demand for cheese production and consumption, have spurred the search for viable alternatives [1, 5]. Various sources of milk-clotting enzymes (MCEs) have been explored, including other animal-derived rennet (from lamb, buffalo, and camel), plant-based coagulants, and microbial and recombinant enzymes. Lamb and buffalo chymosin have lower MCA/PA ratios than calf chymosin, compromising cheese quality, though buffalo rennet’s stability suits mozzarella production [6, 7]. In contrast, camel rennet, while showing desirable clotting properties, is difficult to obtain in large quantities, limiting its widespread use [8]. Rennet from other animals, such as pigs and rabbits, often shows poor specificity and thermal stability, restricting their utility in cheesemaking [1]. Recently, marine organisms such as the shrimp Pleoticus muelleri have also been investigated as novel sources of coagulants [9-11]. Nevertheless, animal-based MCEs continue to face ethical and religious constraints. Plant-based coagulants, such as chymopapain from papaya, offer promising vegan- and halal-friendly alternatives [12]. However, their lower specificity toward κ-casein and tendency to generate bitterness limit their use, especially in aged cheeses [1, 13]. Microbial coagulants, particularly from bacterial and fungal sources, are increasingly favoured due to their cost-effectiveness and suitability for vegetarian and organic cheese production [14]. Although bacterial coagulants offer functional diversity [5, 15-18], they often exhibit high proteolytic activity, necessitating the development of improved strains through mutation [1]. Fungal coagulants are generally more compatible with cheesemaking conditions, particularly in terms of pH and temperature [1, 19]. Still, their high thermostability can cause undesired proteolysis during cheese ripening, adversely affecting texture and flavour [20]. Among fungal coagulants, mucorpepsin (EC 3.4.23.23) from Rhizomucor miehei is widely used due to its relative specificity. However, it still acts on α- and β-caseins and possesses high thermal stability, which can result in residual enzymatic activity after curd formation. This residual activity often leads to bitterness, altered texture, and reduced yield during cheese production [21-25]. Several studies have attempted to address these limitations using physical, chemical, enzymatic, and genetic modifications to enhance specificity, reduce thermostability, and minimise off-target proteolysis while maintaining milk-clotting efficiency. For example, some efforts have successfully reduced thermostability by removing N-linked carbohydrates, one of the primary factors contributing to R. miehei protease’s thermal resistance without significantly diminishing clotting activity [1, 25-29]. Other studies have enhanced milk coagulation without altering thermostability [30-35]. Genetic engineering, mutagenesis [36-39], substrate modification[40-43], and enzyme immobilisation [44, 45], have all been explored to develop efficient coagulants suitable for industrial cheese production. Nevertheless, regulatory restrictions on genetically modified organisms and technical and economic challenges have limited the commercial adoption of many of these approaches [46]. A novel strategy for identifying milk-clotting enzymes involves targeting conserved motifs—namely FDTSSD or FDTGSSE, found in all currently known commercial coagulants. Using this approach, several in silico BLAST searches have identified new candidate proteases for cheese manufacturing. Moreover, degenerate primers based on these motifs can identify relevant genes even when full gene sequences are unknown [26]. Computational enzyme design and molecular modelling have shown great potential in optimizing enzyme activities [47-49]. Advances in bioinformatics, molecular dynamics, and machine learning now enable the prediction and engineering of enzymes with enhanced specificity, catalytic efficiency, and thermostability. Structural insights into active sites support rational design through targeted mutations[50-52]. These in silico methods reduce experimental cost and time by predicting enzyme performance before validation. Notably, ancestral sequence reconstruction has been used to design a novel aspartic protease with improved κ-casein affinity and thermolability for cheesemaking [53]. Although milk-clotting enzymes (MCEs) have been widely studied, the application of advanced computational tools for their rational design remains underexplored [54]. Moreover, current MCEs often exhibit limitations such as low substrate specificity and excessive thermostability, which can negatively affect cheese quality and increase commercial costs. This study introduces a computational pipeline to design thermolabile, κ-casein-specific milk-clotting peptides as sustainable, vegan-, and halal-friendly alternatives for cheese production. An integrated computational pipeline was developed, incorporating machine learning, rational design, and structure-based screening. A combinatorial library of 160,000 peptide variants was generated based on conserved catalytic motifs from the Rhizomucor miehei protease–Pepstatin A complex, linked through a rationally designed sequence. The library was screened using predictive models trained on binding affinity data, followed by structure-based refinement, physicochemical profiling, and validation through molecular docking and molecular dynamics simulations. This in silico framework provides a foundation for the experimental development of next-generation coagulants for dairy applications.
- Materials and Methods
2.1. Aspartic peptide design strategy
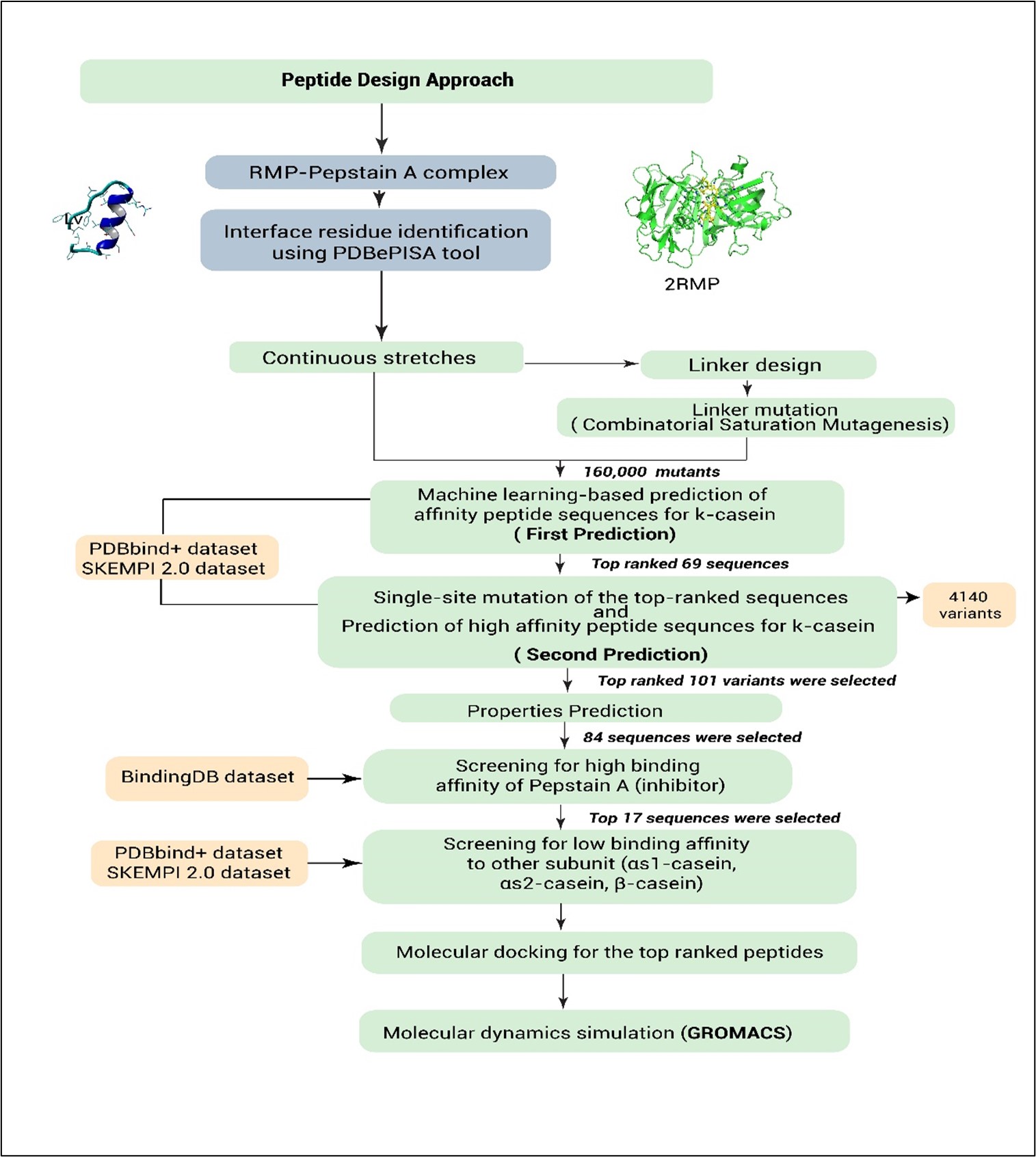
We present a computational pipeline for the de novo design of short peptide-based milk-clotting enzymes (MCEs) with enhanced affinity for κ-casein and improved chemosensitivity. The design strategy involves identifying interface residues containing catalytic motifs from Rhizomucor miehei protease (RMP), linking them via an optimised linker, and screening the resulting peptide candidates for specific binding and functional activity. To benchmark the design process, RMP was used as a positive control and also served as the structural template, while a peptide with low affinity for Pepstatin A was employed as a negative control. The results for both controls are provided in the supplementary file on GitHub.
2.2. Interface residue identification
Interface residues from the crystal structure of 2RMP (RMP–Pepstatin A complex) were identified using the PDBePISA tool (https://www.ebi.ac.uk/pdbe/pisa/) [55] and served as the starting point for aspartic peptidase design. Two continuous interface segments containing the catalytic aspartic acid residues were selected: LFDTGSS (residues 36–42, harbouring the DTGS motif) and TIDTGTNFFI (residues 235–244, containing the DTGT motif). Although the ITYGT segment was identified among the interface residues, it was excluded from the design due to its lack of direct involvement in the conserved DTGS/DTGT catalytic motifs. Furthermore, incorporating non-catalytic segments would unnecessarily increase peptide length without functional contribution, potentially reducing the structural stability and target specificity of the designed peptides.
2.3. Continuous interface residues segment
Catalytic motif of 2RMP (38-41, 237-240)
- LFDTGSS = 36-42 (7 residues)
- TIDTGTNFFI = 235-244 (10 residues)
- ITYGT = 80-84
2.4. Linker design and variant library construction
The designed peptide sequence comprises two continuous interface segments, LFDTGSS (residues 36–42) and TIDTGTNFFI (residues 235–244), totalling 17 residues. The spatial distance between the terminal residues of these segments (Ser42 and Thr235) was measured to be 13.8 Å using chimaera v1.17.3. The average distance between consecutive Cα atoms in a polypeptide chain is approximately 3.8Å [56-58].This distance corresponds to approximately four amino acids, indicating the optimal linker length required to bridge the two segments. Incorporating a four-residue linker (XXXX), the final peptide construct contains 21 amino acids. A saturated mutagenesis approach was applied to the linker, yielding 160,000 variants (20⁴ combinations) of the parent sequence, LFDTGSS-XXXX-TIDTGTNFFI.
2.5.Machine learning models for protein -protein interaction prediction: PDBbind+ and SKEMPI 2.0 Datasets
In this study, datasets were sourced from three major repositories: (1) 160,000 peptide variants generated through combinatorial substitution, (2) 7,086 mutants curated from the SKEMPI 2.0 database, which focuses on mutations in protein–protein interactions[59], and (3) 3,176 protein–protein complexes obtained from the PDBbind+ dataset[60]. For SKEMPI 2.0, duplicates, ambiguous entries (e.g., conflicting binding affinities or incomplete mutation annotations), and incomplete records (e.g., missing sequences or ΔΔG values) were removed. Similarly, the PDBbind+ dataset was filtered by excluding duplicate complexes, sequences with non-canonical residues or extreme lengths, entries lacking binding affinity or structural data, and those with physiologically irrelevant binding values. After filtering, the cleaned data were divided into training (70%), testing (20%), and validation (10%) sets. Two independent machine learning models XGBoost and a Convolutional Neural Network (CNN) were then employed to predict protein–protein interactions.
2.6. SKEMPI 2.0 dataset and CNN-based machine learning model
The SKEMPI 2.0 dataset, comprising 7,086 mutations from 295 studies, was used to train a deep learning model for predicting changes in protein–protein binding affinity[61]. After removing duplicates, ambiguous entries, and incomplete data, 343 high-quality entries were selected, each containing PDB IDs, chain identifiers, affinity values, and protein sequences. Sequences were normalised by peptide length for consistency. Model training was performed using the DeepPurpose framework (v0.1.5)[62], which employs convolutional neural networks (CNNs) for protein sequence encoding. Protein sequences were processed using DeepPurpose’s data_process() function, which internally encodes sequences and applies padding to handle variable lengths without manual preprocessing. The dataset was divided into training (70%), testing (20%), and validation (10%) sets. The model was trained over 100 epochs with a learning rate of 0.001 and a batch size of 32. Performance was evaluated using mean squared error (MSE), R², and Pearson correlation coefficient. The trained model was then used to predict the binding affinity between κ-casein and 160,000 designed peptide variants. Based on predicted scores, the top 21 peptides were selected for further analysis.
2.7. PDBbind+ dataset and XGBoost-based machine learning model
The PDBbind+ dataset, consisting of 3,176 protein–protein complexes [60], was curated by eliminating duplicates, invalid sequences, and missing data, resulting in 1,049 remaining complexes. Affinity values were standardised, log-transformed, and negative values were excluded. A sequence-based predictive model was developed using Conjoint Triad encoding (via DeepPurpose) and logarithmic normalisation to estimate protein–protein and protein–peptide binding affinities. Several machine learning models were tested using Scikit-learn, including Random Forest (R² = 0.67), Ridge (R² = –1.482), Gradient Boosting (R² = 0.647), and XGBoost (R² = 0.696). XGBoost was selected for its superior performance[63]. The model was optimised using the Optuna framework with 100 trials and 5-fold cross-validation, tuning hyperparameters such as estimators (100–1000), learning rate (0.01–0.3), max depth (3–10), subsample (0.6–1.0), and colsample_bytree (0.6–1.0). Outlier detection was performed using an Isolation Forest with a contamination rate of 0.15. The final model, evaluated using mean squared error (MSE), R², and Pearson correlation, was used to predict binding affinities for 160,000 κ-casein peptide variants, identifying the top 48 candidates for further investigation.
2.8. Single-site mutation and machine learning prediction
The top 69 sequences (48 from the model trained on PDBbind+ and 21 from the model trained on SKEMPI 2.0) were further refined through single-site mutations at positions Phe2 (PHE), Ile13 (ILE), and Phe19 (PHE). To preserve peptide structural integrity and avoid disrupting potential hydrogen bonding networks, mutations were excluded from adjacent serine (S) and threonine (T) residues surrounding the linker. Instead, three hydrophobic residues Phe2 (F2), Ile13 (I13), and Phe19 (F19) were carefully chosen as mutation sites due to their roles in local packing and potential linker interactions. Each position was systematically substituted with all 20 canonical amino acids, generating 4,140 unique sequences (20×3×69). The two trained models were used again to predict interactions between these sequences and κ-casein, resulting in 101 selected peptide sequences (96 from the model trained on PDBbind+ and 5 from the model trained on SKEMPI 2.0) with optimal binding affinity.
2.9. Physicochemical property assessment
The top 101 peptide sequences underwent comprehensive screening based on thermal stability, pH, solubility, and toxicity. DeepSTABp (https://csb-deepstabp.bio.rptu.de/) was used to predict melting temperature [64].
ToxinPred (https://webs.iiitd.edu.in/raghava/toxinpred/multi_submit.php) evaluated pH and solubility based on isoelectric point, hydrophobicity, hydrophilicity, amphipathicity, steric hindrance, charge, and molecular weight[65]. ToxinPred2 (https://webs.iiitd.edu.in/raghava/toxinpred2/) [66] and ToxinPred3 (https://webs.iiitd.edu.in/raghava/toxinpred3/index.html) [67] assessed toxicity profiles. This screening identified 84 peptide sequences with favourable physicochemical properties.
2.10. Pepstatin A binding affinity as a proxy for aspartic protease activity
Pepstatin A binding was used as a proxy to assess aspartic protease activity. A pre-trained model, Morgan_CNN_BindingDB_IC50, from the DeepPurpose framework[68], was employed to predict interactions between Pepstatin A and the 84 selected peptides. The model utilised a multi-layer perceptron for drug representation (hidden layers: [1024, 256, 64]) and a convolutional neural network for peptide representation (convolutional layers: filters [32, 64, 96]; kernel sizes [4, 8, 12]). Based on predicted binding affinity, 17 peptides with strong interaction to Pepstatin A were identified for further analysis.
2.11. Cross-reactivity profiling with non-target casein subunits
The 17 selected peptide sequences were further evaluated for binding specificity by assessing their predicted interactions with αs1-casein, αs2-casein, and β-casein, using the same trained models and datasets. Two peptides exhibited low binding affinities to these casein subunits, suggesting a high degree of specificity toward κ-casein. These sequences hold particular promise for targeted applications in unique cheese production processes.
2.12. Predicting and evaluating the 3D structures of the peptidase
The two peptides with the highest binding affinities to Pepstatin A were predicted using PEP-FOLD4 (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD4/) [69], a free tool for predicting linear, cyclic, and chemically modified peptide structures. It uses the improved sOPEP force field and Monte Carlo simulation with Debye-Hückel-free parameters for refinement. The Best Model is based on the lowest SOPEP score. PEP-FOLD4 also provides an SA probability map showing residue conformations: red (helices), green (β-strands), and blue (coils/loops), aiding in stability and secondary structure prediction. MD simulations were run for each peptide, and final stable structures were extracted. Structural quality was then assessed using ERRAT[70], Procheck[71], and verified for further processing.
2.13. Molecular docking of peptidase with κ-casein
Since the X-ray structure of κ-casein remains undetermined, our analysis focused on the local structure surrounding the Phe105–Met106 bond, which is susceptible to aspartic enzyme cleavage in bovine κ-casein. We aimed to explore how this local fragment interacts with RMP and the designed peptides to investigate the underlying mechanism, as highlighted in earlier studies [53, 72, 73]. The same methodology was followed for ligand preparation, with some modifications. The RMP binding site in κ-casein spans residues 102 to 108 [74], while the chymosin binding site covers residues 97 to 112 [75, 76]. The shorter segment (residues 102–108, HLSFMAI) was used as the ligand for docking with RMP, as it corresponds to the P4–P4′ binding region of Pepstatin A in the Co-Crystallised complex with RMP [74], ensuring structural relevance. In contrast, the longer fragment (residues 97–112, RHPHPHLSFMAIPPKK) was used for docking with the designed peptides (Pep 1 and Pep 2) and was also utilised throughout the machine learning pipeline to capture a broader binding context, consistent with its known role in chymosin recognition. This region was predicted using AlphaFold2[77]. Molecular docking was performed using the HADDOCK v2.4-2022.08 web server[78, 79], with the peptides as receptors and κ-casein as the ligand. Asp3 and Asp14 were defined as active residues for the peptides (first molecule), and residues 102 to 108 were set as active for κ-casein (second molecule). The top docking poses were selected based on a combination of criteria, including the size of the largest cluster, the most negative Z-score, the number of hydrogen bonds, and the lowest binding energy. Interaction analyses were carried out using LigPlot+ v1.4.5 software[80].
2.14. Molecular dynamics simulation (MD)
The well-recognised and freely licensed software used in this study was GROMACS version 2024[81]. Topology parameters for the anticipated models were produced using the AMBER force field (ff99SB) in GROMACS [82] .The constructed peptides (pep 1and Pep 2) and RMP models were placed in a cubic box with 10 Å spacing, solvated with the TIP3P water model[83]. Energy minimisation was performed using the steepest descent technique[84], and long-range electrostatics were calculated using the Particle-Mesh Ewald (PME) method[85], with a 1.0 nm cut-off for van der Waals and electrostatic interactions. The maximum number of minimisation steps was 50,000. Equilibration was carried out using 5000 ps of NVT and NPT runs. The V-rescale thermostat (0.1 ps coupling) maintained temperature at 318.15 K, which is the optimal temperature for RMP, during NVT. The Berendsen barostat was employed during the NPT equilibration phase with a coupling time of 2.0 ps to allow fast and stable pressure convergence. Although the Berendsen method does not sample a true NPT ensemble, it is widely used for initial stabilisation [86]. For the subsequent 100 ns production run, the Parrinello–Rahman barostat was used to ensure thermodynamically rigorous sampling of pressure fluctuations and system behaviour [87]. Simulations were also conducted at different temperatures for comparative analysis. Analytical tools including gmx rms, gmx rmsf, gmx gyrate, gmx hbond, and gmx sasa were used to calculate RMSD, RMSF, radius of gyration, hydrogen bonds, and SASA, respectively[88]. All data were extracted from trajectories and visualised using Origin.
2.15. MM-PBSA binding energy, principal component, and free energy landscape analyses of protease complexes
MM-PBSA binding free energy and PCA analyses were conducted using snapshots from the equilibrated MD trajectory at 100 ps intervals. The g_mmpbsa tool was used to calculate the binding free energy (ΔG_bind) based on the equation: ΔGbind = ΔE_MM + ΔG_solvation – TΔS
Where ΔE_MM includes electrostatic and van der Waals interactions, and ΔG_solvation comprises both polar and non-polar solvation energies. The entropy term (TΔS) was approximated or omitted due to computational limitations; therefore, the results mainly represent the enthalpic contribution (ΔH) and reflect approximate binding affinity values. PCA was performed using GROMACS tools (g_covar and g_anaeig) to analyse major motions in the complex based on a mass-weighted covariance matrix of backbone atoms, yielding eigenvectors and eigenvalues that represent motion direction and magnitude[89-91]. Additionally, Free Energy Landscape (FEL) analysis was conducted using RMSD and Radius of Gyration (Rg) as collective variables (CVs), extracted via gmx rms and gmx gyrate. The FEL was calculated using the Boltzmann relation, ΔG = −k_BT ln P(CV1, CV2), and visualised as a 2D contour map using gmx sham, highlighting stable (low-energy) and unstable (high-energy) conformational states [92].
2.16. Statistical analysis
Quantitative data analysis was performed using GraphPad Prism 9, with results presented as mean ± standard deviation. A one-way analysis of variance (ANOVA) was used to evaluate differences across groups, followed by Tukey's multiple comparisons test to identify specific pairwise differences. Statistical significance was set at p < 0.05.
- Results and Discussion
3.1. First prediction: ML-based affinity screening of peptides targeting κ-casein
We configured DeepPurpose to process our peptide dataset (160,000) using two tailored machine learning models trained on the SKEMPI 2.0 and PDBbind+ databases, with both datasets split into 70% training, 20% testing, and 10% validation sets. For the PDBbind+ model, we applied an XGBoost-based algorithm utilizing Conjoint Triad encoding and logarithmic normalisation. Outliers were removed using Isolation Forest (contamination = 0.15). This model achieved a Pearson correlation coefficient of 0.8360, an R² value of 0.6907, and a mean squared error (MSE) of 116. 4807. In contrast, the SKEMPI 2.0 model was constructed using a convolutional neural network (CNN) within the DeepPurpose framework, with input sequences normalised by length. The model architecture included hidden layers of 64 and 32 units, a learning rate of 0.001, a batch size of 32, and training over 100 epochs. It yielded stronger predictive performance, with a Pearson correlation of 0.964, R² of 0.92, and MSE of 0.0022. Following the prediction, 69 top peptide sequences were selected 48 from the PDBbind+ model and 21 from the SKEMPI 2.0 model for further evaluation against κ-casein. The SKEMPI-derived sequences showed variable predicted binding scores, ranging from 0.124221 to 0.1120, reflecting nuanced differences in predicted affinities captured by the CNN (Fig. 1). In contrast, all PDBbind+-derived sequences received an identical predicted score of –8.08991, potentially due to structural similarity among candidates or model limitations in resolving fine-grained differences. The dual-model approach enabled complementary evaluation of peptide binding predictions. The CNN-based model trained on SKEMPI 2.0 exhibited superior predictive accuracy, likely due to its ability to learn fine-grained local patterns relevant to short peptide interactions. This aligns with prior studies showing that convolutional architectures are well-suited for capturing local dependencies in biological sequences and outperform tree-based methods in small, mutation-sensitive datasets such as SKEMPI2.0 [93, 94]. The performance metrics suggest that this model captured subtle sequence-affinity relationships better than the XGBoost model trained on PDBbind+, which may be less sensitive to small mutations and optimised for larger, structurally diverse protein–protein interfaces[95]. These findings underscore the critical influence of dataset composition, sequence representation, and algorithmic architecture in machine learning–guided protein design. By integrating two complementary models, the current investigation enhances confidence in candidate selection and provides a framework for future predictive workflows in rational peptide engineering.
3.2. Second prediction: Single-site mutagenesis and ML-based optimisation of peptide affinity for κ-casein
To improve κ-casein binding, 4,140 single-site variants were generated from 69 lead peptides, each following the structure LFDTGSSXXXXTIDTGTNFFI. To maintain peptide structure and hydrogen bonding, mutations near serine (S) and threonine (T) residues flanking the linker were avoided. Instead, three hydrophobic residues, Phe2 (F2), Ile13 (I13), and Phe19 (F19) involved in local packing and linker interactions were selected for mutation. Each was independently substituted with all 20 canonical amino acids. The variants were evaluated using machine learning models trained on PDBbind+ and SKEMPI 2.0 datasets. From this second prediction, 101 top candidates were selected: 96 from the PDBbind+ model (all with a predicted score of 1.3525908) and 5 from SKEMPI 2.0 (scores ranging from 0.062 to 0.088) (Fig. 2). The identical scores from PDBbind+ are likely due to the XGBoost algorithm combined with a descriptor set that captures general physicochemical properties. This descriptor likely assigned similar feature values to many peptides, resulting in minimal differentiation in predicted affinity. This highlights both the utility and the limitations of such models, particularly when applied to datasets with structurally and chemically similar variants.
3.3. Assessment of physicochemical properties
Comprehensive evaluation of physicochemical properties is critical in the selection of functional peptides for industrial and biomedical applications, particularly in processes such as milk coagulation. Where solubility, thermal sensitivity, and non-toxicity are essential traits. In this study, 101 computationally selected peptides were screened using in silico tools to predict their toxicity, charge distribution, solubility, molecular weight, and thermal stability. ToxinPred analysis confirmed that all peptides were non-toxic, with negative SVM scores between -1.03 and -0.24, which is consistent with safety requirements for food-grade peptide additives[65]. These findings minimise concerns about cytotoxic or allergenic effects, thereby supporting their potential use in dairy fermentation systems. The net charge of most peptides was -3, and their isoelectric points (pI) ranged from 3.57 to 4.21, indicating strong acidity and that they remain negatively charged under physiological conditions. This negative charge may promote electrostatic repulsion between peptide molecules, reducing aggregation and enhancing aqueous solubility—an important attribute for homogeneous distribution in milk matrices[96]. The peptides’ hydrophilic nature, inferred from their sequence composition, and moderate molecular weights (2225–2590 Da), also suggest favourable pharmacokinetic behaviour and low aggregation potential, features typically associated with improved stability and bioavailability [97]. Thermal stability, as predicted by DeepSTABp, showed melting temperatures ranging from 55.27°C to 59.59°C. These moderate thermal profiles are advantageous for milk-clotting enzymes, as excessive thermal stability may hinder enzyme deactivation post-clotting, while overly heat-labile peptides might degrade during processing [46]. Thus, the predicted melting temperatures reflect an optimal balance for heat-sensitive processes, potentially allowing efficient clotting at typical curdling temperatures (around 40–45°C) while avoiding residual activity that could affect cheese texture during ageing. Integrating these diverse parameters—non-toxicity, net negative charge, solubility, appropriate molecular weight, and moderate thermostability—allowed the rational narrowing of the peptide pool to 84 candidates. These peptides represent promising leads for development as aspartic protease-like milk coagulants, offering a non-animal alternative with tunable biochemical properties tailored for dairy industry needs.
3.4. Binding selectivity analysis
3.4.1. Aspartic Activity Analysis: Binding Affinity of Peptides Toward Pepstatin A
As part of our strategy to design peptides that functionally mimic natural aspartic proteases, we used predicted binding affinity to Pepstatin A, a specific inhibitor of aspartic proteases, as a proxy for enzymatic activity. Peptides with high predicted affinity are likely to exhibit aspartic protease-like functionality, justifying their use in functional screening. To assess this, 84 peptides were evaluated for their binding affinity to Pepstatin A using the Morgan_CNN_BindingDB_IC50 model (Fig. 3A). This model was selected due to its strong methodological and biological relevance: it is trained on BindingDB, which contains extensive IC₅₀ data for enzyme–inhibitor interactions, including those involving aspartic proteases. The incorporation of Morgan fingerprints enables effective representation of small molecules such as Pepstatin A, while the convolutional neural network (CNN) architecture facilitates the extraction of features pertinent to molecular binding. Among the models tested, this one demonstrated superior performance in predicting small-molecule–protein interactions. Given Pepstatin A’s role as a potent and structurally well-defined inhibitor of aspartic proteases, the model is particularly suitable for estimating binding affinities in this context. The model, trained over 800 iterations, demonstrated rapid convergence, with loss values decreasing from 0.24 to near zero. Validation metrics indicated high predictive performance (R² = 0.8277, MSE = 0.0025, Pearson r = 0.98) (Fig. 3B). Predicted binding affinities ranged from 5.325 to 5.240 (log IC₅₀), suggesting consistently strong interactions. These findings support the functional aspartic activity of the peptides and their potential utility in cheese-making applications. The top 17 peptides were selected for further analysis (Fig. 3C). The use of the Morgan_CNN_BindingDB_IC50 model, a deep learning-based approach trained on the SKEMPI 2.0 dataset, allowed for high-fidelity prediction of molecular interactions, as evidenced by robust performance metrics (R² = 0.8277, MSE = 0.0025, Pearson r = 0.98) [98, 99]. The model’s rapid convergence and low final loss further highlight its reliability in capturing relevant biochemical features governing binding behaviour. The narrow range of predicted binding affinities (log IC₅₀ values between 5.325 and 5.240) suggests a high degree of consistency among the peptides in terms of their interaction strength with Pepstatin A. This indicates that many of the evaluated peptides may possess conserved structural or functional motifs typical of aspartic proteases, aligning with the hypothesis of their functional relevance. Such strong predicted interactions imply not only potential inhibitory characteristics but also functional mimicry of natural aspartic proteases, which are integral to milk clotting and protein breakdown in cheese production.
3.4.2. Cross-reactivity evaluation with non-target casein subunits
To further evaluate the specificity of the designed peptides, the top 17 sequences with strong predicted binding affinity to Pepstatin A were analysed for cross-reactivity against non-target casein subunits (αs1-, αs2-, and β-casein) using the same machine learning model (Fig. 4). The average cross-reactivity score among these peptides was calculated to be 0.431529 (Table 1). Based on this threshold, six peptides with cross-reactivity values below the average were selected as more selective candidates. Among them, Pep1 (LFDTGSSVDEMTRDTGTNFFI; log IC₅₀ = 5.325; Cross-Reactivity = 0.3636) and Pep2 (LFDTGSSADESTRDTGTNFFI; log IC₅₀ = 5.309; Cross-Reactivity = 0.2706) showed both the lowest cross-reactivity scores and high predicted binding affinity to Pepstatin A. To assess structural reliability, all 17 peptides were subjected to molecular dynamics (MD) simulations. The root-mean-square deviation (RMSD) of backbone atoms was used as an indicator of conformational stability. Pep1 exhibited the lowest RMSD value (0.407 nm), followed closely by Pep2 (0.513 nm), indicating that both peptides maintain stable conformations during simulation and are structurally reliable (Fig. 5). These combined criteria, low cross-reactivity, high binding affinity, and strong conformational stability were used to identify Pep1 and Pep2 as the top candidates for κ-casein-specific targeting in cheese-making applications. Pep1 showed a slightly stronger interaction with Pepstatin A (5.325). Pep2 exhibited the lowest cross-reactivity with all non-target caseins, particularly αs2-casein (0.0717), indicating higher substrate specificity. These results suggest that Pep1 is better suited for applications requiring high catalytic efficiency. Whereas Pep2 is ideal for precise selectivity in cheese-making processes. These findings underscore the importance of balancing catalytic potency with substrate specificity when designing casein-targeting peptides for dairy applications[100]. Although Pep1 demonstrates superior binding affinity to Pepstatin A, its slightly higher predicted interactions with αs-caseins could pose a risk of off-target proteolysis during cheese processing [101]. In contrast, Pep2’s exceptionally low cross-reactivity enhances its utility for applications where minimal disruption of non-κ-casein proteins is critical, such as in the production of speciality cheeses with defined texture and flavour profiles [102]. Future in vitro and in situ validations will be essential to confirm these in silico predictions and to optimise the application of these peptides in industrial cheese-making protocols. Overall, this approach highlights the potential of machine learning-guided screening for rational peptide design in food biotechnology [103] .
3.5. Structure prediction and validation
The structural validation of the designed peptides was carried out using PEP-FOLD 4 modelling, Ramachandran plot analysis, and ERRAT evaluation to ensure their stereochemical quality and overall stability. For the first designed peptide (Pep1), the predicted 3D structure generated by PEP-FOLD demonstrated a well-folded and compact conformation, indicating a plausible structural model (Fig. 6A). The use of PEP-FOLD 4, a well-established de novo modelling tool, facilitates the generation of compact and reliable peptide conformations. It enables the identification of energetically favourable structures that mimic biologically relevant folds, which is critical during early-stage peptide design [69]. The Ramachandran plot showed that 94.1% of the residues were located in the most favoured regions, with the remaining 5.9% distributed in the additional allowed regions (Fig 6B). This distribution suggests that nearly all backbone dihedral angles fall within sterically permissible regions, reflecting a highly acceptable stereochemical quality. Such a high percentage of residues in the most favoured regions is a hallmark of high-resolution protein structures and indicates minimal strain in backbone geometry [71]. Moreover, the ERRAT quality factor for this peptide was 100%, indicating an excellent profile for non-bonded atomic interactions and confirming the high reliability of the predicted structure (Fig. 6C). The ERRAT score of 100% indicates that the model has non-bonded atomic interactions comparable to those found in well-refined experimental structures, further reinforcing its structural reliability [70]. Similarly, for the second designed peptide (Pep2), the structure predicted by PEP-FOLD also revealed a stable conformation consistent with a biologically plausible fold (Fig. 6D). The recurrence of a compact and stable fold across multiple models supports the reproducibility and robustness of the peptide design strategy. The Ramachandran plot analysis mirrored the results observed for the first peptide, with 94.1% of residues positioned in the most favoured regions and 5.9% in additional allowed regions (Fig. 6E). This consistency reinforces the structural integrity of the designed peptide and highlights the effectiveness of the design strategy. It also suggests that the second peptide maintains favourable backbone torsion angles and avoids sterically hindered conformations, indicating a structurally sound model [104] The ERRAT analysis for the second peptide also yielded a perfect score of 100%, demonstrating the presence of highly favourable atomic interactions comparable to those found in high-resolution crystal structures (Fig. 6F). This high ERRAT value underscores the accuracy of atomic-level geometry and supports the overall reliability of the structural prediction for Pep2. The data exhibited that both designed peptides Pep1 and Pep2 successfully met important structural quality standards. The large number of amino acids in preferred areas of the Ramachandran plot, along with excellent ERRAT scores, strongly suggests that the predicted structures are sound and of high quality. The agreement between both stereochemical and atomic-level validation methods strongly supports the robustness of the design and predictive workflow. Such consistency not only strengthens confidence in the structural integrity of the peptides but also indicates their suitability for further studies involving interaction modelling, stability profiling, or therapeutic evaluation. This indicates that the peptides have correct stereochemistry and form stable, dependable three-dimensional shapes, which suggests they could be useful for more research into their structure, function, or potential as therapies.
3.6. Molecular docking analyses for Pep1 and Pep2
Docking analysis was conducted to assess the binding affinity and interaction profiles of the two designed peptides (Pep 1 and Pep 2) in comparison with the reference molecule RMP (2ASI). Several key parameters were evaluated, including Z-score, docking score, number of hydrogen bonds, salt bridges, hydrophobic interactions, and cleavage sites. In the HADDOCK docking results, Pep 1 exhibited the lowest Z-score of -1.7, indicating the most energetically favourable and structurally reliable model among the three ligands. Pep 2 followed with a Z-score of -1.5, while the reference RMP had the highest Z-score of -1.1, suggesting comparatively less stable binding. These findings imply that both designed peptides adopt favourable conformations within the receptor's binding site, with Pep 1 demonstrating the most promising structural quality. In terms of HADDOCK scoring, where more negative values suggest more favourable predicted interactions, Pep 1 achieved the best score among the peptides (-78.9 ± 0.1), followed by Pep 2 (-69.5 ± 1.2). The reference full-length enzyme RMP exhibited a more negative score (-126 ± 7.5), likely due to its larger size (360 amino acids) and broader interaction surface, which allows for more extensive electrostatic and van der Waals interactions. Therefore, direct comparison between short peptides (21 amino acids) and the full-length enzyme should be interpreted with caution, as HADDOCK scores are influenced by molecular size and interaction complexity rather than binding strength alone. Nonetheless, the favourable scores of Pep 1 and Pep 2 indicate that these peptides can form stable and biologically relevant interactions with the target receptor. Their interaction energies fall within the range typically observed for biologically active peptides in similar docking studies, supporting their potential as viable aspartic protease candidates [105]. Interaction profiling further revealed that Pep 1 formed 9 hydrogen bonds and 15 hydrophobic contacts (Fig. 7A), Pep 2 established 13 hydrogen bonds and 11 hydrophobic contacts (Fig. 7B). The formation of multiple hydrogen bonds, especially in Pep 2, implies strong polar interactions that can enhance specificity and complex stability [106]. In contrast, the higher hydrophobic interaction count in Pep 1 may promote tighter binding through van der Waals forces and nonpolar surface compatibility [107] . The absence of salt bridges suggests that electrostatic interactions are not the primary mode of binding in this system, a feature shared by many short bioactive peptides.
The higher number of hydrogen bonds in Pep 2 suggests stronger polar interactions, potentially contributing to its specific binding orientation and stability. The hydrophobic interaction count was higher in Pep 1, which might enhance the overall binding affinity through nonpolar surface contacts. Regarding cleavage specificity, Pep 1 was cleaved at a phenylalanine-methionine (F-M) site, whereas both Pep 2 and RMP shared cleavage at a histidine (H) site, which might reflect a similarity in their interaction pattern with the proteolytic target. This similarity in cleavage site between Pep 2 and RMP might suggest a conserved interaction motif or susceptibility pattern, which could be explored further in functional assays [108]. The unique cleavage site in Pep 1, however, could imply a distinct mechanism of interaction or processing. Both designed peptides displayed favourable binding characteristics, with Pep 1 showing stronger hydrophobic interactions and Pep 2 demonstrating more hydrogen bonding. Although the docking score of the reference molecule was considerably higher, the designed peptides showed stable interactions and energetically favourable profiles, indicating their potential suitability as functional analogs or inhibitors with acceptable structural and binding quality. Generally speaking, the docking results suggest that both Pep 1 and Pep 2 exhibit promising interaction profiles, with complementary binding features Pep 1 emphasising hydrophobic engagement and Pep 2 leveraging polar contacts. Such dual characteristics offer flexibility for future optimisation strategies, either for enhancing potency or tailoring specificity toward target modulation. These findings support further investigation of both peptides in experimental settings for potential therapeutic or diagnostic applications.
3.7. Structural and thermostability of the designed Pep1
To evaluate the structural stability of the designed peptide Pep1 under varying thermal conditions, molecular dynamics (MD) simulations were conducted at five different temperatures (30 °C, 40 °C, 45 °C, 50 °C, and 60 °C) over a 100 ns period. Analyses included root-mean-square deviation (RMSD), solvent accessible surface area (SASA), radius of gyration (Rg), intramolecular hydrogen bonding, and root-mean-square fluctuation (RMSF) to comprehensively assess the peptide's conformational behaviour. RMSD analysis showed Pep1’s highest stability at 40°C (0.3–0.4 nm), with minimal structural deviation, while higher temperatures increased instability. At 30 °C and 45 °C, the RMSD values remained below 0.6 nm, suggesting overall stability with some minor structural adjustments. However, at 50 °C, RMSD values rose to approximately 0.7 nm, signifying partial destabilisation. The most pronounced structural deviations were observed at 60 °C, where RMSD frequently exceeded 1.0 nm and approached 1.5 nm, indicative of significant conformational changes and potential unfolding (Fig. 8A). This pattern reflects how increasing temperature leads to progressive structural disruption, as RMSD serves as a sensitive global indicator of backbone deviation from the native fold [109]. Stable RMSD at 40 °C suggests a thermodynamically favourable conformation, while higher RMSD at 50–60 °C may result from weakened non-covalent interactions and increased backbone flexibility, typical of thermally induced unfolding. SASA analysis supported these observations by providing insight into solvent exposure across temperatures. Throughout the 100 ns simulations, SASA values generally ranged from 20 to 27 nm². At 40 °C and 45 °C, the peptide maintained relatively consistent and moderate SASA values, reflecting a compact and well-folded structure. At 30 °C, the SASA profile remained fairly stable, though transient spikes suggested brief conformational openings. In contrast, simulations at 50 °C and 60 °C showed elevated and fluctuating SASA values, signifying increased solvent exposure likely due to structural expansion or partial unfolding (Fig. 8B). Since SASA quantifies the surface area of the peptide accessible to solvent molecules, lower SASA is typically associated with buried hydrophobic cores and tighter packing [110]. The moderate values at 40 °C confirm an optimal fold, whereas increasing SASA at higher temperatures implies exposure of internal residues, suggesting disruption of hydrophobic interactions and unfolding behaviour under thermal stress. The radius of gyration (Rg) further illustrated temperature-induced changes in compactness. At 40 °C, Pep1 exhibited the most stable and lowest Rg values, reinforcing the inference of a tightly packed conformation. Rg values at 45 °C remained close to those at 40 °C with minor fluctuations, suggesting sustained compactness. At 30 °C, intermittent increases in Rg were observed, reflecting occasional conformational relaxation. Conversely, at 50 °C and 60 °C, pronounced increases and fluctuations in Rg indicated a loss of compactness and a tendency toward structural loosening and partial unfolding (Fig. 8C). The radius of gyration is a geometric measure reflecting the peptide’s mass distribution around its center. Stable and low Rg values at 40 °C are characteristic of folded states [111], while rising Rg at 50–60 °C implies a structural transition toward more extended, unfolded forms. These findings align with literature reports where thermally induced unfolding is marked by increased Rg due to chain expansion.
Hydrogen bond analysis revealed a direct relationship between temperature and the internal cohesion of the peptide. The highest and most consistent number of hydrogen bonds—ranging between 15 and 22—was observed at 30 °C and 40 °C, indicating strong intra-peptide interactions and a stable secondary structure. At 45 °C, the hydrogen bond count remained stable but showed increased fluctuations. Significantly reduced and highly variable hydrogen bonding was seen at 50 °C, while at 60 °C, the number of hydrogen bonds frequently dropped below 10 and occasionally below 5, reflecting substantial destabilisation and disruption of the peptide’s internal architecture (Fig. 8D). Intramolecular hydrogen bonds are crucial for maintaining the secondary structure of peptides, particularly α-helices and β-turns. Their reduction at high temperatures supports the unfolding hypothesis, as these bonds are sensitive to thermal fluctuations [112]. The drop below 10 hydrogen bonds at 60 °C suggests substantial loss of secondary structural elements, consistent with peptide denaturation phenomena observed in other MD-based peptide studies. RMSF analysis provided residue-level insights into flexibility across the peptide sequence. At 40 °C, Pep1 demonstrated the lowest RMSF values across nearly all residues, particularly in the central core (residues 6–15), indicating localised rigidity and structural conservation. RMSF values increased gradually at 45 °C and 50 °C, while the most pronounced fluctuations were observed at 60 °C, especially at the N- and C-terminal regions. These heightened fluctuations at elevated temperatures suggest increased dynamic motion and structural instability under thermal stress (Fig. 8E). RMSF reveals the flexibility of individual residues. Low RMSF in the core region of Pep1 at 40 °C implies a well-constrained, rigid fold—critical for maintaining functional conformation. The rising RMSF at termini under heat stress is typical, as unstructured terminal regions tend to destabilise first, acting as 'thermal sensors' during unfolding [113].
Together, these comprehensive analyses underscore that 40 °C is the most favourable temperature for maintaining the structural stability and compactness of Pep1. At this temperature, Pep1 exhibits minimal deviation, low solvent exposure, strong hydrogen bonding, compact geometry, and limited flexibility hallmarks of a stable, functional conformation, further supported by a favourable binding free energy (MM/PBSA: -50.2 kcal/mol). In contrast, higher temperatures, particularly 50 °C and 60 °C, induce progressive structural destabilisation, likely compromising the peptide's structural integrity and potential biological activity. To further evaluate thermostability, the TM Predictor web server (http://tm.life.nthu.edu.tw) was used, yielding a predicted thermostability index (TI) of -4.05 for Pep1, indicating reduced thermal stability compared to the reference RMP, which exhibited a higher TI of 0.1688. The enzyme with the lower TI value is expected to melt first, as a lower TI reflects reduced thermostability. The average structural and energetic properties of Pep 1 under various temperature conditions, derived from MD simulations, are summarised in Table 2.
Structural and thermostability of the designed Pep2
To comprehensively assess the temperature-dependent stability and structural dynamics of the designed peptide Pep2, all-atom molecular dynamics (MD) simulations were performed at 30 °C, 40 °C, 45 °C, 50 °C, and 60 °C for 100 ns. A suite of structural metrics—including root mean square deviation (RMSD), solvent-accessible surface area (SASA), radius of gyration (Rg), intramolecular hydrogen bonding, and root mean square fluctuation (RMSF)—was evaluated. The results are presented below in a structured order, integrating both quantitative findings and mechanistic interpretations. Pep2 displayed the lowest and most stable RMSD values (0.2–0.6 nm) at 40 °C, indicating minimal conformational deviation and a highly stable backbone over the simulation period. At 30 °C and 45 °C, RMSD values increased modestly (0.4–0.8 nm), suggesting partial flexibility or incipient destabilisation. More pronounced fluctuations were observed at 50 °C (~0.9 nm), and at 60 °C, RMSD peaked at ~1.4 nm, signifying substantial structural deviation and partial unfolding. RMSD data indicate that 40 °C is the optimal thermal condition for structural integrity. The relatively low deviations at this temperature reflect a well-preserved conformation with sufficient thermal energy to promote natural flexibility without destabilisation. At 30 °C, limited thermal motion likely reduces conformational sampling, slightly increasing rigidity. Higher temperatures disrupt stabilizing interactions and increase molecular motion, explaining the substantial deviation at 60 °C. These findings align with typical protein folding behaviour, where a narrow thermal window supports optimal native-like stability (Fig. 9A).
At 40 °C, Pep2 showed the lowest SASA values (15–25 nm²), indicative of a compact, well-folded conformation with limited solvent interaction. At 30 °C and 45 °C, SASA values were moderately higher, suggesting less optimal folding. At elevated temperatures (50 °C and 60 °C), SASA increased markedly (~27–28 nm²), consistent with partial unfolding and exposure of internal residues to the solvent. The minimised SASA at 40 °C is consistent with effective hydrophobic core packing and maintenance of tertiary structure. As temperature rises, increased kinetic energy leads to expansion and loosening of the structure, exposing previously buried hydrophilic and hydrophobic residues. The increase in SASA at 60 °C particularly reflects unfolding events. Conversely, the moderate SASA at 30 °C suggests compactness but possibly over-constrained structural rigidity.
These results reinforce 40 °C as the most favourable temperature for maintaining folding efficiency and native-like solvation characteristics (Fig. 9B). Rg analysis confirmed a tightly packed structure at 40 °C, with values ranging between 0.75 and 0.9 nm and minimal fluctuation. At 30 °C, similar compactness was observed but with slightly increased variation. At 45 °C, Rg variability increased and exhibited a noticeable spike (~1.4 nm) near 80 ns, indicating transient expansion. At 50 °C and 60 °C, Rg values rose steadily (up to ~1.2 nm), signaling loss of compactness. Rg trends corroborate the conclusions drawn from RMSD and SASA. The tight distribution at 40 °C reflects optimal intramolecular packing, possibly due to a well-preserved hydrophobic core and favourable enthalpic interactions. The Rg increase at higher temperatures is indicative of thermal-induced expansion, likely driven by disruption of non-covalent stabilizing forces. The transient Rg spike at 45 °C suggests an intermediate destabilisation point, while sustained Rg elevation at 60 °C reflects a transition toward a partially denatured state (Fig. 9C).
Pep2 maintained the highest number of hydrogen bonds at 40 °C (17–22), indicating strong internal cohesion. At 30 °C, values remained relatively high (15–18) but were more variable. The number of hydrogen bonds declined with increasing temperature: 10–15 at 45–50 °C and dramatically reduced to 0–12 at 60 °C (Fig. 9D). Hydrogen bonding plays a critical role in stabilizing both secondary and tertiary structural elements. The high number of stable hydrogen bonds at 40 °C strongly supports the peptide's compact, folded state, as seen in RMSD and Rg data. The variability at 30 °C may result from reduced structural dynamics, limiting optimal hydrogen bond formation. Elevated temperatures likely disrupt hydrogen bonds through increased molecular motion and thermal agitation, leading to unfolding and instability, particularly at 60 °C. These results validate hydrogen bonding as a key contributor to Pep2’s thermal resilience.
RMSF values were lowest at 40 °C across nearly all residues, especially within the central core, indicating reduced flexibility and a stable fold. At 30 °C and 50 °C, moderate fluctuations (0.3–0.6 nm) were observed. At 45 °C and 60 °C, RMSF values increased notably, particularly at the N- and C-termini, with fluctuations reaching ~0.8 nm at 60 °C (Fig. 9E). Residue-level flexibility is a crucial indicator of structural adaptability and stability. The low RMSF at 40 °C reflects an optimally stabilized structure with minimal disorder, especially in regions typically prone to fluctuation. Elevated RMSF at higher temperatures points to local unfolding and increased mobility, beginning at the termini and potentially propagating inward. At 30 °C, limited thermal energy may suppress flexibility, potentially compromising functional dynamics. These observations underscore the role of dynamic balance in protein stability where both excessive rigidity and excessive motion can be detrimental.
The comprehensive MD analysis of Pep2 at varying temperatures identifies 40 °C as the optimal thermal condition for structural stability, compactness, and internal cohesion. At this temperature, the peptide maintains low RMSD and SASA, minimal Rg, a robust hydrogen bond network, and reduced local flexibility indicators of a well-folded, functionally favourable conformation, further supported by a binding free energy of -39.07 kcal/mol (MM/PBSA). Both suboptimal (30 °C) and elevated (45–60 °C) temperatures compromise one or more structural features, culminating in significant destabilisation at 60 °C. These insights are critical for the rational design and application of Pep2 in biotechnological settings where thermal resilience and functional integrity are required. Supporting this observation, the TM Predictor estimated a thermostability index (TI) of -3.09 for Pep2. The average structural and energetic properties of Pep 2 under various temperature conditions, derived from MD simulations, are summarised in Table 3.
3.8. Free energy landscape for designed Pep1 and Pep2
3.8.1. FEL analyses of Pep1
Based on the free energy landscape (FEL) plots provided for the Pep1–κ-casein complex at temperatures 30°C (Fig. 10A), 40°C (Fig. 10B), 45°C (Fig. 10C), 50°C (Fig. 10D), and 60°C (Fig. 10E), the conformational dynamics and binding stabilities vary significantly across temperatures. At 30°C, the basin is located at CV1 = –10, CV2 = 5, indicating a relatively deep and well-defined minimum in terms of free energy (in kJ/mol), suggesting a stable conformational state for the complex. At 40°C, the basin shifts to CV1 = 20, CV2 = 10, representing a higher energy state (kJ/mol) and broader basin, implying increased molecular fluctuations and possibly a less stable binding. At 45°C, the basin center is at CV1 = 10, CV2 = 0, with a more moderate energy well (kJ/mol), indicating a transiently stable interaction. At 50°C (CV1 = –10, CV2 = –10) and 60°C (CV1 = 0, CV2 = –10), the landscapes display multiple shallow basins or broader minima, again in kJ/mol, suggesting conformational heterogeneity and possible unfolding or weakened binding interactions. Comparing all FELs, 30°C shows the most localised and lowest energy basin (in kJ/mol), indicating that it offers the most favourable condition for stable Pep1–κ-casein interaction. Thus, 30°C appears to be the optimal temperature for strong and stable binding between the protein and ligand, as higher temperatures lead to increased conformational entropy and destabilisation of the complex. In ligand–peptide systems, FELs provide insight into the dynamic ensemble of bound states and the thermodynamic landscape that governs molecular recognition. The single deep basin observed at 30°C suggests a dominant, energetically favourable binding conformation with limited flexibility, implying a strong and specific interaction. In contrast, the emergence of multiple basins at elevated temperatures indicates increased conformational sampling and reduced energy barriers between alternative states, which often corresponds to transient or weak binding modes and reduced specificity [114]. Such behaviour is characteristic of thermally induced disruptions in ligand anchoring, possibly due to peptide backbone fluctuations or loss of key interaction contacts. These results highlight the temperature sensitivity of the Pep1–κ-casein interface and underscore the utility of FELs in visualising binding plasticity and thermodynamic preferences in peptide-ligand complexes.
3.8.2. FEL analyses of Pep2
The Free Energy Landscape (FEL) plots of the Pep2–κ-casein complex across five temperatures (30°C to 60°C) reveal distinct thermodynamic behaviours and conformational shifts, as represented by the collective variables (CV1 and CV2) and their corresponding energy minima in kJ/mol. At 30°C (Fig. 11A), three basins are evident. The global minimum is located at CV1 = –10 and CV2 = 0 with the lowest free energy (~0 kJ/mol), indicating a highly stable conformation of the Pep2–κ-casein complex. Additionally, two lighter blue basins at slightly higher CV coordinates (~CV1 = 0 to 5 and CV2 = 5–10) represent metastable conformations within 3–5 kJ/mol of the global minimum. The presence of multiple low-energy basins suggests that at 30°C, the complex not only adopts its most stable structure but also maintains conformational flexibility, which is often beneficial for functional protein–ligand interactions (e.g., induced fit or dynamic binding modes). At 40°C (Fig. 11B), the global minimum shifts slightly upward to CV1 = –10 and CV2 = 5, still maintaining a deep and compact energy basin (~1–2 kJ/mol), indicative of a still-stable complex. However, the landscape becomes marginally less diverse, suggesting slightly reduced conformational flexibility compared to 30°C. At 45°C (Fig. 11C), a dramatic conformational shift is observed with the basin center at CV1 = 10 and CV2 = 10. The basin is shallower, with energy increasing to ~5–6 kJ/mol, and the landscape appears more diffuse. This suggests that the complex is adopting a different, less favourable binding mode, likely due to thermal destabilisation. The reduced depth and increased spread of the basin imply both lower thermodynamic stability and higher structural fluctuations, which may compromise binding affinity or functional specificity. At 50°C (Fig .11D), the basin moves to CV1 = 10 and CV2 = 0, with slightly better stability than at 45°C but still higher free energy than at 30–40°C (~4–5 kJ/mol).
The loss of distinct, multiple basins indicates reduced structural adaptability, pointing toward the beginning of a denaturation trend or significant conformational compromise. Finally, at 60°C (Fig. 11E), the basin shifts to CV1 = –10 and CV2 = 15, but the energy is notably higher (~7–8 kJ/mol), and the landscape is broad and poorly defined. This suggests the complex is significantly destabilised, with increased conformational entropy, weakened interactions, and probable unfolding or loss of functional binding. In summary, 30°C provides the most favourable thermodynamic conditions for Pep2–κ-casein interaction, with the lowest free energy basin (~0 kJ/mol) and the presence of additional shallow basins (~3–5 kJ/mol) indicating conformational flexibility and stability. 40°C remains acceptable, albeit with slightly less flexibility. Temperatures above 45°C led to altered binding modes and increased free energies, reducing stability and functional interaction potential. Therefore, based on FEL analysis, 30°C is the optimal temperature for the stable and flexible binding of Pep2 to κ-casein. Molecular mechanics Poisson–Boltzmann surface area (MM/PBSA) calculations indicated that the lowest average binding free energy (ΔG_binding) for the Pep2–κ-casein complex occurred at 40 °C. As MM/PBSA directly estimates the binding affinity by evaluating the enthalpic contributions (electrostatic, van der Waals, and solvation energies) from molecular dynamics snapshots, this result suggests that 40 °C provides the most energetically favourable condition for static peptide–protein interactions. In contrast, free energy landscape (FEL) analysis, which evaluates the thermodynamic distribution of conformational states over time using collective variables (e.g., RMSD, radius of gyration), revealed that 30 °C is associated with the deepest global free energy minimum. This indicates that at 30 °C, the complex adopts a more thermodynamically stable and dynamically accessible conformation. Unlike MM/PBSA, FEL does not directly quantify binding affinity but instead captures the stability and accessibility of the conformational ensemble sampled during the simulation. The discrepancy between the two analyses highlights the complementary nature of these methods: MM/PBSA reflects the strength of the final bound interaction (affinity), while FEL provides insight into the conformational thermodynamics and stability of the binding process. Taken together, these results suggest that while 40 °C may yield the strongest static binding affinity, 30 °C may represent a more favourable temperature for achieving a stable and functionally relevant protein–ligand complex due to enhanced conformational sampling and thermodynamic stability.
3.9. Principal component analyses of Pep1 and Pep2
3.9.1. PCA analyses of Pep1
Principal component analysis (PCA) was conducted to evaluate the conformational behaviour of the Pep1–κ-casein complex under increasing thermal conditions (30–60°C). At 30°C (Fig. 12A), the trajectory shows multiple well-defined and tightly packed clusters, suggesting the presence of distinct and stable conformational states with limited structural deviation — indicative of strong and specific interactions between Pep1 and κ-casein at physiological temperature. As the temperature increases to 40°C (Fig. 12B) and 45°C (Fig. 12C), the clustering pattern becomes more dispersed and less compact, indicating enhanced molecular flexibility and the beginning of a shift toward less stable binding conformations. By 50°C (Fig. 12D), the PCA projection reveals fewer and more diffuse clusters with longer inter-cluster transitions, reflecting a pronounced increase in structural variability and possible weakening of the peptide–protein interaction. At 60°C (Fig. 12E), the trajectory exhibits highly scattered points with minimal recurrent clustering, suggesting the breakdown of stable conformations and potentially partial or full dissociation of the complex. This temperature-dependent trajectory dispersion reflects thermal destabilisation, where higher kinetic energy leads to disruption of specific contacts and structural rearrangement within the complex. These findings demonstrate that the Pep1–κ-casein interaction is thermosensitive, with optimal stability occurring at or below 30–40°C, while elevated temperatures progressively disrupt the conformational landscape, reducing the likelihood of functional binding interactions. This PCA-based analysis indicated the crucial role of temperature in modulating ligand–protein interactions. The increasing conformational heterogeneity at higher temperatures indicates a reduction in binding affinity and stability, as the thermal motion overcomes non-covalent interactions, such as hydrogen bonds and hydrophobic contacts that stabilize the complex [115]. Similar trends have been observed in other peptide–protein systems, where elevated temperatures lead to the loss of native binding conformations and functional disruption [116]. Therefore, the progressive dispersion of the PCA clusters can be interpreted as a signature of thermal denaturation and interaction loss, reinforcing the necessity to consider physiological temperature constraints in the design and application of bioactive peptide therapeutics.
3.9.2. PCA analyses of Pep2
Principal Component Analysis (PCA) was conducted to investigate the temperature-dependent conformational dynamics of the interaction between the designed Pep2 and κ-casein, providing insights into the stability and flexibility of the complex across five different temperatures. At 30 °C (Fig. 13A), the PCA plot displayed a compact and highly clustered trajectory distribution, indicating limited atomic fluctuations and a rigid conformational space, reflective of a highly stable and well-structured Pep2–κ-casein complex. This suggests that at lower temperatures, the complex maintains structural integrity with minimal conformational shifts. At 40 °C (Fig. 13B), the trajectory began to show a slightly broader dispersion, though still relatively cohesive, suggesting moderate flexibility that may facilitate conformational accommodation without compromising binding stability.
This moderate adaptability could enhance interaction specificity and affinity by enabling Pep2 to better conform to κ-casein’s surface. At 45 °C (Fig. 13C), the PCA plot revealed multiple loosely defined clusters, indicating increased conformational sampling and transitions among distinct substates. Such dynamic behaviour suggests a threshold temperature where the complex begins to lose rigidity, potentially leading to reduced binding affinity or altered interaction profiles. A pronounced shift was observed at 50 °C (Fig. 13D), where the distribution became highly scattered and lacked a dominant conformational basin, signifying significant conformational fluctuations and loss of structural coherence. This degree of flexibility points to a destabilised interaction, with the possibility of partial unfolding or loss of binding contacts. At 60 °C (Fig. 13E), the trajectory remained widely dispersed, though with a slightly directional pattern, indicating large-scale conformational motions likely associated with denaturation or dissociation events. Collectively, these findings suggest that Pep2 maintains a stable and functionally relevant interaction with κ-casein up to 40 °C. However, as the temperature increases beyond 45 °C, the complex exhibits increasing conformational plasticity and instability, which may impair functional binding and structural integrity. The PCA analysis underscores the critical influence of temperature on the dynamic behaviour of protein–peptide complexes and highlights 30–40 °C as the optimal thermal window for Pep2–κ-casein interaction stabilityTaken together, both Pep1 and Pep2 demonstrated strong clotting activity at 40 °C and maintained structural stability within this moderate temperature range, whereas the control enzyme RMP showed optimal activity at 45 °C. This lower optimal temperature, combined with their high specificity, makes Pep1 and Pep2 better suited for industrial cheesemaking processes that favour moderate thermal conditions, potentially enhancing energy efficiency and process control. These features offer significant advantages for a variety of soft and hard cheeses that coagulate optimally under these conditions. Moreover, as animal rennet alternatives, Pep1 and Pep2 provide precise coagulation control essential for all cheese formulations, including vegan and halal varieties. Additionally, they are well-suited for cost-effective, large-scale production using microbial expression systems such as E. coli or Pichia pastoris, enabling scalable and food-grade manufacturing. Table 4 summarises the comparative features specifically, specificity, binding affinity, and thermolability highlighting the advantages of Pep1 and Pep2 over the control enzyme RMP for commercial cheese production.
3.10. Validation approach and future directions
To support the computational findings and address the inherent limitations of in silico approaches, future work will involve synthesising the top candidate peptides, Pep1 and Pep2, and experimentally evaluating their milk-clotting activity under controlled laboratory conditions. While computational modelling provides valuable insights into peptide performance, it has several limitations: it relies on theoretical force fields, may not fully account for post-translational modifications or folding dynamics, and cannot replicate the full complexity of biological systems such as enzyme–substrate interactions in milk matrices. These factors highlight the necessity of empirical validation. Pep1 and Pep2 will be produced recombinantly in Escherichia coli or Pichia pastoris and then purified. The peptides will subsequently undergo in vitro assays to assess milk-clotting activity, specificity toward κ-casein, and thermal sensitivity, with tests conducted at 30–40 °C to mimic industrial cheesemaking conditions. Additionally, thermostability and pH profiles will be evaluated to benchmark performance against commercial microbial coagulants. These experimental validations will be essential to confirm our computational predictions and advance the peptides’ potential for industrial application.
- Conclusion
This study presents the rational design of peptide-based biocatalysts and addresses longstanding challenges in cheese biotechnology particularly the demand for ethical, specific, and thermolabile milk-clotting enzymes. By integrating interface residue engineering, machine learning-based prediction, and molecular dynamics (MD) simulations, we established a scalable and reproducible framework for designing milk-clotting peptides. Among the designed candidates, Pep1 and Pep2 outperformed existing milk-clotting enzymes (MCEs), with optimal activity predicted at 30–40 °C. These characteristics align well with the operational conditions of industrial cheesemaking. While the results are promising, experimental validation remains necessary. Overall, this computational pipeline represents a meaningful advancement in enzyme design for dairy applications.
- Acknowledgements
The authors would like to acknowledge the support from the Biotechnology and Biobased Industry Research Program, Universiti Putra Malaysia, 43400 Serdang, Selangor, Malaysia.
This research was supported by the Fundamental Research Grant Scheme (FRGS) from the Ministry of Higher Education (MoHE), Malaysia (FRGS/1/2024/STG05/UPM/02/11), awarded to the last author (SNO).
Data Availability:
All underlying data and scripts are publicly available at: https://github.com/shilansaleem/Aspartic-Peptidases. The results of both positive and negative controls are also available on GitHub. Additional data will be made available upon reasonable request.
- Declaration of competing interest
The authors declare no competing interests.
- Authors’ Contributions
SSS, SNO, and MBAR conceived the study, designed the methodology, and wrote the manuscript. SSS conducted the research. OMA, ATCL, ABS, and NDMN verified the data.
- Using Artificial Intelligent Chatbots
No AI chatbots or tools were used in this research in data analysis, scientific content generation or interpretation.
- Ethical Consideration
This article does not contain any studies involving human participants or animals performed by any of the authors.
- Milk-clotting enzymes
- Computational peptide design
- κ-casein specificity
- Thermolabile peptides
- Sustainable cheese production
- Molecular dynamics simulations
- Machine learning
- Vegan cheese production
- Halal cheese production
- Aspartic protease
How to Cite
References
Liu X, Wu Y, Guan R, Jia G, Ma Y, Zhang Y. Advances in research on calf rennet substitutes and their effects on cheese quality. Food Res Int. 2021; 149: 110704. https://doi.org/10.1016/j.foodres.2021.110704
Nicosia FD, Pino A, Maciel GL, Sanfilippo RR, Caggia C, de Carvalho AF, Randazzo CL. Technological characterisation of lactic acid bacteria strains for potential use in cheese manufacture. Foods. 2023;12(6): 1154. https://doi.org/10.3390/foods12061154
Di Rosa AR, Accetta F, Nicosia FD, Litrenta F, Pino A, Lopreiato V, Caggia C, Randazzo CL. Microbiological, chemical, and artificial sensory assessment of Sicilian cheeses made using different milk-clotting enzymes. Food Biosci. 2024; 59: 103917. https://doi.org/10.1016/j.fbio.2024.103917
Jacob M, Jaros D, Rohm H. Recent advances in milk clotting enzymes. Int J Dairy Technol. 2011;64(1): 14-33. https://doi.org/10.1111/j.1471-0307.2010.00633.x
Wehaidy HR, Abdel-Naby MA, Kholif AM, Elaaser M, Bahgaat WK, Wahab WA. The catalytic and kinetic characterization of Bacillus subtilis MK775302 milk clotting enzyme: Comparison with calf rennet as a coagulant in white soft cheese manufacture. J Genet Eng Biotechnol. 2023; 21(1): 61. https://doi.org/10.1186/s43141-023-00513-w
Vacca GM, Stocco G, Dettori ML, Bittante G, Pazzola M. Goat cheese yield and recovery of fat, protein, and total solids in curd are affected by milk coagulation properties. J Dairy Sci. 2020; 103(2): 1352-1365. https://doi.org/10.3168/jds.2019-16424
Sales DC, Rangel AH, Urbano SA, Tonhati H, Galvão JG, Guilhermino MM, Aguiar EM, Bezerra MD. Buffalo milk composition, processing factors, whey constituents recovery and yield in manufacturing Mozzarella cheese. Food Sci Technol. 2017; 38(2): 328-334. https://doi.org/10.1590/1678-457X.04317
Langholm Jensen J, Mølgaard A, Navarro Poulsen JC, Harboe MK, Simonsen JB, Lorentzen AM, Hjernø K, van den Brink JM, Qvist KB, Larsen S. Camel and bovine chymosin: The relationship between their structures and cheese-making properties. Acta Crystallogr D Biol Crystallogr. 2013; 69(Pt 5): 901-913. https://doi.org/10.1107/S0907444913003260
Pereira ND, Fernández-Gimenez AV. Exogenous enzymes in dairy technology: Acidic proteases from processing discards of shrimp Pleoticus muelleri and their use as milk-clotting enzymes for cheese manufacture. Int J Food Sci Technol. 2017; 52(2): 341-347. https://doi.org/10.1111/ijfs.13285
Rossano R, Larocca M, Riccio P. Digestive enzymes of the crustaceans Munida and their application in cheese manufacturing: A review. Mar Drugs. 2011; 9(7): 1220-1231. https://doi.org/10.3390/md9071220
Shamsuzzaman K, Haard NF. Evaluation of harp seal gastric protease as a rennet substitute for Cheddar cheese. J Food Sci. 1983 Jan;48(1):179-82. https://doi.org/10.1111/j.1365-2621.1983.tb14818.x
Duarte AR, Duarte DM, Moreira KA, Cavalcanti MT, Lima-Filho JL, Porto AL. Jacaratia corumbensis O. Kuntze: A new vegetable source for milk-clotting enzymes. Braz Arch Biol Technol. 2009; 52: 1-9. https://doi.org/10.1590/S1516-89132009000100001
Liu J, Sharma A, Niewiara MJ, Singh R, Ming R, Yu Q. Papain-like cysteine proteases in Carica papaya: Lineage-specific gene duplication and expansion. BMC Genomics. 2018;19(1): 26. https://doi.org/10.1186/s12864-017-4394-y
Neef J, Bongiorni C, Schmidt B, Goosens VJ, van Dijl JM. Relative contributions of non-essential Sec pathway components and cell envelope-associated proteases to high-level enzyme secretion by Bacillus subtilis. Microb Cell Fact. 2020; 19(1): 52. https://doi.org/10.1186/s12934-020-01315-2
Guleria S, Walia A, Chauhan A, Shirkot CK. Optimisation of milk-clotting enzyme production by Bacillus amyloliquefaciens SP1 isolated from apple rhizosphere. Bioresour Bioprocess. 2016; 3(1): 30. https://doi.org/10.1186/s40643-016-0108-6
Meng F, Chen R, Zhu X, Lu Y, Nie T, Lu F, Lu Z. Newly effective milk-clotting enzyme from Bacillus subtilis and its application in cheese making. J Agric Food Chem. 2018 ; 66(24): 6162–6169. https://doi.org/10.1021/acs.jafc.8b01697
Zhang Y, Xia Y, Ding Z, Lai PF, Wang G, Xiong Z, Liu X, Ai L. Purification and characteristics of a new milk-clotting enzyme from Bacillus licheniformis BL312. LWT. 2019; 113: 108276. https://doi.org/10.1016/j.lwt.2019.108276
Wehaidy HR, Abdel-Naby MA, Shousha WG, Elmallah MI, Shawky MM. Improving the catalytic, kinetic and thermodynamic properties of Bacillus subtilis KU710517 milk clotting enzyme via conjugation with polyethylene glycol. Int J Biol Macromol. 2018; 111: 296–301. https://doi.org/10.1016/j.ijbiomac.2017.12.125
Addis M, Piredda G, Pirisi A. The use of lamb rennet paste in traditional sheep milk cheese production. Small Rumin Res. 2008; 79(1): 2–10. https://doi.org/10.1016/j.smallrumres.2008.07.002
Mamo J, Kangwa M, Fernandez-Lahore HM, Assefa F. Optimisation of media composition and growth conditions for production of milk-clotting protease (MCP) from Aspergillus oryzae DRDFS13 under solid-state fermentation. Braz J Microbiol. 2020; 51(2): 571–584. https://doi.org/10.1007/s42770-020-00243-y
Kheirolomoom A. Optimisation of mucor miehei rennin production and recovery. Sci Iran. 2002; 9(1).
Aljammas HA, Al Fathi H, Alkhalaf W. Study the influence of culture conditions on rennin production by Rhizomucor miehei using solid-state fermentations. J Genet Eng Biotechnol. 2018; 16(1): 213-216. https://doi.org/10.1016/j.jgeb.2017.10.004
Escobar J, Barnett SM. Effect of agitation speed on the synthesis of Mucor miehei acid protease. Enzyme Microb Technol. 1993; 15(12): 1009-1013. https://doi.org/10.1016/0141-0229(93)90047-6
Tiwari BD. Microbial protein and its application in cheese making. In: Indian Council of Agricultural Research, editor. Application of biotechnology in dairy and food processing. Karnal: Indian Council of Agricultural Research; 2003. p. 179–183
Leite Júnior BR, Kubo MT, Augusto PE, Atribst AA. High-pressure homogenization on food enzymes. In: Knoerzer K, Muthukumarappan K, editors. Innovative Food Processing Technologies: A Comprehensive Review. 1st ed. Cambridge: Woodhead Publishing; 2021. p. 421–428. https://doi.org/10.1016/B978-0-08-100596-5.22999-5
Feijoo Siota L, Blasco L, Rodríguez Rama JL, Barros Velázquez J, de Miguel T, Sánchez Pérez A, Villa TG. Recent patents on microbial proteases for the dairy industry. Recent Adv DNA Gene Seq. 2014; 8(1): 44–55. https://doi.org/10.2174/2352092208666141013231720
Brown ED, Yada RY. A kinetic and equilibrium study of the denaturation of aspartic proteinases from the fungi Endothia parasitica and Mucor miehei. Biochim Biophys Acta. 1991; 1076(3): 406–415. https://doi.org/10.1016/0167-4838(91)90484-H
Branner-Jorgensen S. Thermal destabilisation of microbial rennet. US Patent 4,255,454. 1981 Mar 10.
Cornelius DA, inventor; Miles Laboratories Inc, assignee. Process for decreasing the thermal stability of microbial rennet. US Patent 4,348,482. 1982 Sep 7
Harboe MK. Rhizomucor miehei aspartic proteinases having improved properties. In: James MNG, editor. Advances in Experimental Medicine and Biology. Boston, MA: Springer US; 1998. p. 293–296. https://doi.org/10.1007/978-1-4615-5373-1_40
Harada K, Kobayashi H, Suga T, Yamaguchi H, Tsunoda A, Kato S, inventors; Meito Sangyo KK, assignee. Milk-clotting protease derived from a microorganism. US Patent 8,609,389. 2013 Dec 17
Aljammas HA, Yazji S, Azizieh A. Enhancement of protease production from Rhizomucor miehei by mutagenesis with ethyl methanesulfonate, ultraviolet, and microwaves—A preliminary study. Bioresour Technol Rep. 2022; 20: 101287. https://doi.org/10.1016/j.biteb.2022.101287
Cornelius DA, Asmus CV, Sternberg MM, inventors; Miles Laboratories Inc, assignee. Acylation of Mucor pusillus microbial rennet enzyme. US Patent 4,362,818. 1982 Dec 7.
Harboe MK, Kristensen PB, inventors; Chr Hansen AS, assignee. Microbially derived enzymes having enhanced milk clotting activity and method of producing same. US Patent 6,127,142. 2000 Oct 3
Júnior BR, Tribst AA, Cristianini M. Influence of high pressure homogenization on commercial protease from Rhizomucor miehei: Effects on proteolytic and milk clotting activities. LWT Food Sci Technol. 2015; 63(1): 739–744. https://doi.org/10.1016/j.lwt.2015.03.020
Feijoo-Siota L, Rama JL, Sánchez-Pérez A, Villa TG. Expression, activation and processing of a novel plant milk-clotting aspartic protease in Pichia pastoris. J Biotechnol. 2018; 268: 28-39. https://doi.org/10.1016/j.jbiotec.2018.01.006
Badiefar L, Ahmadian G, Asgarani E, Ghandili S, Salek Esfahani M, Khodabandeh M. Optimisation of conditions for expression and activation of a splice variant of prochymosin lacking exon 6 in Escherichia coli. Int J Dairy Technol. 2009; 62(2): 265–271. https://doi.org/10.1111/j.1471-0307.2009.00474.x
Suzuki J, Hamu A, Nishiyama M, Horinouchi S, Beppu T. Site-directed mutagenesis reveals functional contribution of Thr218, Lys220 and Asp304 in chymosin. Protein Eng. 1990; 4(1): 69–71. https://doi.org/10.1093/protein/4.1.69
Lahteenmaki L, Grunert K, Ueland O, Astrom A, Arvola A, Bech-Larsen T. Acceptability of genetically modified cheese presented as real product alternative. Food Qual Prefer. 2002; 13(7-8): 523-533. https://doi.org/10.1016/S0950-3293(01)00077-5
Visser S, Slangen CJ, van Rooijen PJ. Peptide substrates for chymosin (rennin). Interaction sites in κ-casein-related sequences located outside the (103-108)-hexapeptide region that fits into the enzyme's active-site cleft. Biochem J. 1987; 244(3): 553-558. https://doi.org/10.1042/bj2440553
Visser S, Van Rooijen PJ, Slangen CJ. Peptide substrates for chymosin (rennin) isolation and substrate behaviour of two tryptic fragments of bovine ϰ casein. Eur J Biochem. 1980; 108(2): 415-421. https://doi.org/10.1111/j.1432-1033.1980.tb04737.x
Raymond MN, Garnier J, Bricas E, Cilianu S, Blasnic M, Chaix A, Lefrancier P. Studies on the specificity of chymosin (rennin): I-Kinetic parameters of the hydrolysis of synthetic oligopeptide substrates. Biochimie. 1972; 54(2): 145-154. https://doi.org/10.1016/S0300-9084(72)80098-1
Morihara K, Hiroshige T. Comparative study of various neutral proteinases from microorganisms: Specificity with oligopeptides. Arch Biochem Biophys. 1971; 146(1): 291-296. https://doi.org/10.1016/S0003-9861(71)80066-8
Channe PS, Shewale JG. Continuous production of cheese by immobilised milk-clotting protease from Aspergillus niger MC4. Biotechnol Prog. 1998; 14(6): 885-889. https://doi.org/10.1021/bp980092k
Al Awwaly KU. Immobilization of rennin enzyme from Mucor pusillus with alginate and its application in cheese making. J Indones Trop Anim Agric. 2007; 32(4): 222-229.
Qasim F, Diercks-Horn S, Gerlach D, Schneider A, Fernandez-Lahore HM. Production of a novel milk-clotting enzyme from solid-substrate Mucor spp. Culture. J Food Sci. 2022; 87(10): 4348-4362. https://doi.org/10.1111/1750-3841.16307
Dušan P, Lynn KS. Molecular modelling of conformational dynamics and its role in enzyme evolution. Curr Opin Struct Biol. 2018; 52: 50-57. https://doi.org/10.1016/j.sbi.2018.08.004
Musil M, Konegger H, Hon J, Bednar D, Damborsky J. Computational design of stable and soluble biocatalysts. ACS Catal. 2018; 9(2): 1033-1054. https://doi.org/10.1021/acscatal.8b03613
Dinmukhamed T, Huang Z, Liu Y, Lv X, Li J, Du G, Liu L. Current advances in design and engineering strategies of industrial enzymes. Syst Microbiol Biomanuf. 2021; 1(1): 15-23. https://doi.org/10.1007/s43393-020-00005-9
Nair VS, George JJ. Computational approaches for protein stability studies. Recent Trends Sci Technol. 2018; 51-56. https://doi.org/10.5281/zenodo.4727263
Ming Y, Wang W, Yin R, Zeng M, Tang L, Tang S, Li M. A review of enzyme design in catalytic stability by artificial intelligence. Brief Bioinform. 2023; 24(3): bbad065. https://doi.org/10.1093/bib/bbad065
Naveed M, Nadeem F, Mehmood T, Bilal M, Anwar Z, Amjad F. Protease—A versatile and co-friendly biocatalyst with multi-industrial applications: An updated review. Catal Lett. 2021; 151(2): 307-323. https://doi.org/10.1007/s10562-020-03316-7
Saleem SS, Akinola OM, Rahman MB, Leow AT, Noor ND, Salleh AB, Oslan SN. Designing novel aspartic protease (NAP) using ancestral sequence reconstruction (ASR) for cheesemaking. Mol Biotechnol. 2025; 1-23. https://doi.org/10.1007/s12033-025-01470-0
Theron LW, Divol B. Microbial aspartic proteases: Current and potential applications in industry. Appl Microbiol Biotechnol. 2014; 98(21): 8853-8868. https://doi.org/10.1007/s00253-014-6035-6
Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007; 372(3): 774-797. https://doi.org/10.1016/j.jmb.2007.05.022
Branden CI, Tooze J. Introduction to Protein Structure. 2nd ed. New York: Garland Science; 2012 https://doi.org/10.1201/9781136969898
Chothia C. Structural invariants in protein folding. Nature.1975; 254(5498): 304-308. https://doi.org/10.1038/254304a0
Creighton TE. Proteins: Structures and Molecular Properties. 2nd ed. New York: W.H. Freeman; 1993.
Jankauskaitė J, Jimenez-Garcia B, Dapkunas J, Fernandez-Recio J, Moal IH. SKEMPI 2.0: An updated benchmark of changes in protein–Protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics. 2019; 35(3): 462-469. https://doi.org/10.1093/bioinformatics/bty635
Wang R, Fang X, Lu Y, Yang CY, Wang S. The PDBbind database: Methodologies and updates. J Med Chem; 48(12): 4111-4119. https://doi.org/10.1021/jm048957q
Jankauskaitė J, Jiménez-García B, Dapkūnas J, Fernández-Recio J, Moal IH. SKEMPI 2.0: An updated benchmark of changes in protein–Protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics. 2019; 35(3): 462-469. https://doi.org/10.1093/bioinformatics/bty635
Hashemifar S, Neyshabur B, Khan AA, Xu J. Predicting protein–Protein interactions through sequence-based deep learning. Bioinformatics. 2018; 34(17): i802-810. https://doi.org/10.1093/bioinformatics/bty573
Chen T, Guestrin C. XGBoost: A scalable tree boosting system. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; 2016 Aug 13–17; San Francisco, CA, USA. New York: ACM; 2016. p. 785-794. https://doi.org/10.1145/2939672.2939785
Jung F, Frey K, Zimmer D, Mühlhaus T. DeepSTABp: A deep learning approach for the prediction of thermal protein stability. Int J Mol Sci. 2023; 24(8): 7444. https://doi.org/10.3390/ijms24087444
Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Open-Source Drug Discovery Consortium, Raghava GP. In silico approach for predicting toxicity of peptides and proteins. PLoS One. 2013; 8(9): e73957. https://doi.org/10.1371/journal.pone.0073957
Sharma N, Naorem LD, Jain S, Raghava GP. ToxinPred2: An improved method for predicting toxicity of proteins. Brief Bioinform. 2022; 23(5): bbac174. https://doi.org/10.1093/bib/bbac174
Rathore AS, Choudhury S, Arora A, Tijare P, Raghava GP. ToxinPred 3.0: An improved method for predicting the toxicity of peptides. Comput Biol Med. 2024; 179: 108926. https://doi.org/10.1016/j.compbiomed.2024.108926
Huang K, Fu T, Glass LM, Zitnik M, Xiao C, Sun J. DeepPurpose: A deep learning library for drug–target interaction prediction. Bioinformatics. 2020;36(22-23): 5545-5547. https://doi.org/10.1093/bioinformatics/btaa1005
Rey J, Murail S, de Vries S, Derreumaux P, Tuffery P. PEP-FOLD4: A pH-dependent force field for peptide structure prediction in aqueous solution. Nucleic Acids Res. 2023; 51(W1): W432-W437. https://doi.org/10.1093/nar/gkad376
Colovos C, Yeates TO. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993; 2(9): 1511-1519. https://doi.org/10.1002/pro.5560020916
Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993; 26(2): 283-291. https://doi.org/10.1107/S0021889892009944
Li T, Wang J, Li Y, Zhang L, Zheng L, Li Z, Yang Z, Luo Q. Structure of the complex between Mucor pusillus pepsin and the key domain of κ-casein for site-directed mutagenesis: A combined molecular modelling and docking approach. J Mol Model. 2011; 17(7): 1661-1668. https://doi.org/10.1007/s00894-010-0869-3
Zhang J, Sun Y, Li Z, Luo Q, Li T, Wang T. Structure-based design of Mucor pusillus pepsin for the improved ratio of clotting activity/proteolytic activity in cheese manufacture. Protein Pept Lett. 2015; 22(7): 660-667. https://doi.org/10.2174/0929866522666150526154648
Yang J, Teplyakov A, Quail JW. Crystal structure of the aspartic proteinase from Rhizomucor miehei at 2.15 Å resolution. J Mol Biol. 1997; 268(2): 449-459. https://doi.org/10.1006/jmbi.1997.0968
Sørensen J, Palmer DS, Qvist KB, Schiøtt B. Initial stage of cheese production: A molecular modeling study of bovine and camel chymosin complexed with peptides from the chymosin-sensitive region of κ-casein. J Agric Food Chem. 2011; 59(10): 5636-5647. https://doi.org/10.1021/jf104898w
Sørensen J, Palmer DS, Schiøtt B. Hot-spot mapping of the interactions between chymosin and bovine κ-casein. J Agric Food Chem. 2013; 61(33): 7949-7959. https://doi.org/10.1021/jf4021043
Bryant P, Pozzati G, Elofsson A. Improved prediction of protein-protein interactions using AlphaFold2. Nat Commun. 2022; 13(1): 1265. https://doi.org/10.1038/s41467-022-28865-w
Dominguez C, Boelens R, Bonvin AM. HADDOCK: A protein−Protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003; 125(7): 1731-1737. https://doi.org/10.1021/ja026939x
van Zundert GC, Rodrigues JP, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond AS, van Dijk M, de Vries SJ, Bonvin AM. The HADDOCK2.2 web server: user-friendly integrative modelling of biomolecular complexes. J Mol Biol. 2016; 428(4): 720-725. https://doi.org/10.1016/j.jmb.2015.09.014
Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng Des Sel. 1995; 8(2): 127-134. https://doi.org/10.1093/protein/8.2.127
Alekseenko A, Páll S, Lindahl E. GROMACS on AMD GPU-based HPC platforms: Using SYCL for performance and portability. arXiv [preprint]. 2024; arXiv: 2405.01420. Available from: https://doi.org/10.48550/arXiv.2405.01420
Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006; 65(3): 712–725. https://doi.org/10.1002/prot.21123
Han N, Miao H, Yu T, Xu B, Yang Y, Wu Q, Zhang R, Huang Z. Enhancing thermal tolerance of Aspergillus niger PhyA phytase directed by structural comparison and computational simulation. BMC Biotechnol. 2018; 18(1): 36. https://doi.org/10.1186/s12896-018-0445-y
Huang L, Ma J, Sang J, Wang N, Wang S, Wang C, et al. Enhancing the thermostability of phospholipase D from Streptomyces halstedii by directed evolution and elucidating the mechanism of a key amino acid residue using molecular dynamics simulation. Int J Biol Macromol. 2020; 164: 3065-3074. https://doi.org/10.1016/j.ijbiomac.2020.08.160
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577–8593. https://doi.org/10.1063/1.470117
Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984; 81(8):3684–3690. https://doi.org/10.1063/1.448118
Parrinello M, Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys. 1981; 52(12): 7182–7190. https://doi.org/10.1063/1.328693
Hess B, Kutzner C, Van Der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008; 4(3): 435-447. https://doi.org/10.1021/ct700301q
Singh JK, Anand S, Srivastava SK. Is BF. 7 more infectious than other Omicron subtypes: Insights from structural and simulation studies of BF. 7 spike RBD variant. Int J Biol Macromol. 2023; 238: 124154. https://doi.org/10.1016/j.ijbiomac.2023.124154
David CC, Jacobs DJ. Principal component analysis: A method for determining the essential dynamics of proteins. In: Tobias DJ, editor. Protein Dynamics: Methods and Protocols. Methods Mol Biol. New York: Humana Press; 2014. p. 193-226. https://doi.org/10.1007/978-1-62703-658-0_11
Sultana, K. N., & Srivastava, S. K. Structural and molecular dynamics of ammonia transport in Staphylococcus aureus NH3-dependent NAD synthetase. Int J Biol Macromol. 2022; 203: 593–600. https://doi.org/10.1016/j.ijbiomac.2022.01.138
Baidya, A. T., Kumar, A., Kumar, R., & Darreh-Shori, T. Allosteric binding sites of Aβ peptides on the acetylcholine synthesising enzyme ChAT as deduced by in silico molecular modelling. Int J Mol Sci. 2022; 23(11): 6073. https://doi.org/10.3390/ijms23116073
Zhang, C., Sun, Y., & Hu, P. An interpretable deep geometric learning model to predict the effects of mutations on protein–protein interactions using large-scale protein language model. J Cheminform. 2025; 17(1): 35. https://doi.org/10.1186/s13321-025-00979-5
Çevrim E, Yiğit MG, Ulusoy E, Yılmaz A, Doğan T. A benchmarking platform for assessing protein language models on function-related prediction tasks. bioRxiv [preprint]. 2025. Available from: https://doi.org/10.1101/2025.04.10.648084
Khan S, Noor S, Javed T, Naseem A, Aslam F, AlQahtani SA, Ahmad N. XGBoost-enhanced ensemble model using discriminative hybrid features for the prediction of sumoylation sites. BioData Mining. 2025; 18(1): 12. https://doi.org/10.1186/s13040-024-00415-8
Kramer RM, Shende VR, Motl N, Pace CN, Scholtz JM. Toward a molecular understanding of protein solubility: Increased negative surface charge correlates with increased solubility. Biophys J. 2012; 102(8): 1907-1915. https://doi.org/10.1016/j.bpj.2012.01.060
Gomes B, Augusto MT, Felício MR, Hollmann A, Franco OL, Gonçalves S, Santos NC. Designing improved active peptides for therapeutic approaches against infectious diseases. Biotechnol Adv. 2018; 36(2): 415-429. https://doi.org/10.1016/j.biotechadv.2018.01.004
Moal IH, Fernández-Recio J. SKEMPI: A structural kinetic and energetic database of mutant protein interactions and its use in empirical models. Bioinformatics. 2012; 28(20): 2600-2607. https://doi.org/10.1093/bioinformatics/bts489
Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007; 35(Database issue): D198-201. https://doi.org/10.1093/nar/gkl999
Korhonen H, Pihlanto A. Bioactive peptides: Production and functionality. Int Dairy J. 2006; 16(9): 945-960. https://doi.org/10.1016/j.idairyj.2005.10.012
Gobbetti M, Stepaniak L, De Angelis M, Corsetti A, Di Cagno R. Latent bioactive peptides in milk proteins: Proteolytic activation and significance in dairy processing. Crit Rev Food Sci Nutr. 2002; 42(3): 223-239. https://doi.org/10.1080/10408690290825538
McSweeney PL, Sousa MJ. Biochemical pathways for the production of flavour compounds in cheeses during ripening: A review. Le Lait. 2000; 80(3): 293-324. https://doi.org/10.1051/lait:2000127
Mohanty DP, Mohapatra S, Misra S, Sahu DP. Milk derived bioactive peptides and their impact on human health–A review. Saudi J Biol Sci. 2016; 23(5): 577–583. https://doi.org/10.1016/j.sjbs.2015.06.005
Sobolev OV, Afonine PV, Moriarty NW, Hekkelman ML, Joosten RP, Perrakis A, Adams PD. A global Ramachandran score identifies protein structures with unlikely stereochemistry. Structure. 2020; 28(11):1249–1258. https://doi.org/10.1016/j.str.2020.08.005
Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat Rev Drug Disco. 2004; 3(11): 935–949. https://doi.org/10.1038/nrd1549
Nezhad NG, Abd Rahman RN, Normi YM, Oslan SN, Shariff FM, Leow TC. Recent advances in simultaneous thermostability-activity improvement of industrial enzymes through structure modification. Int J Biol Macromol. 2023; 232: 123440. https://doi.org/10.1016/j.ijbiomac.2023.123440
Nezhad NG, Buhari SB, Eskandari A, Albayati SH, Omotayo OF, Leow TC. Improving the thermostability and substrate affinity of IsPETase through the S209H mutation, and exploring the structural role of the N-terminal: A computational study. 3 Biotech. 2025; 15(4): 93. https://doi.org/10.1007/s13205-025-04258-w
Moriguchi T, Haraguchi K, Ueda N, Okada M, Furuya T, Akiyama T. DREG, a developmentally regulated G protein‐coupled receptor containing two conserved proteolytic cleavage sites. Genes to Cells. 2004; 9(6): 549-560. https://doi.org/10.1111/j.1356-9597.2004.00743.x
Kurniawan J, Ishida T. Protein model quality estimation using molecular dynamics simulation. ACS Omega. 2022; 7(28): 24274-24281. https://doi.org/10.1021/acsomega.2c01475
Qing R, Hao S, Smorodina E, Jin D, Zalevsky A, Zhang S. Protein design: From the aspect of water solubility and stability. Chem Rev. 2022; 122(18): 14085-14179. https://doi.org/10.1021/acs.chemrev.1c00757
Benrezkallah D. Molecular dynamics simulations at high temperatures of the Aeropyrum pernix L7Ae thermostable protein: Insight into the unfolding pathway. J Mol Graph Model. 2024; 127: 108700. https://doi.org/10.1016/j.jmgm.2023.108700
Arnittali M, Rissanou AN, Amprazi M, Kokkinidis M, Harmandaris V. Structure and thermal stability of wtRop and RM6 proteins through all-atom molecular dynamics simulations and experiments. Int J Mol Sci. 2021; 22(11): 5931. https://doi.org/10.3390/ijms22115931
Medvedev KE, Alemasov NA, Vorobjev YN, Boldyreva EV, Kolchanov NA, Afonnikov DA. Molecular dynamics simulations of the Nip7 proteins from the marine deep-and shallow-water Pyrococcus species. BMC Struct Biol. 2014; 14(1): 23. http://www.biomedcentral.com/1472-6807/14/23
Abouzied AS, Alqarni S, Younes KM, Alanazi SM, Alrsheed DM, Alhathal RK, Huwaimel B, Elkashlan AM. Structural and free energy landscape analysis for the discovery of antiviral compounds targeting the cap-binding domain of influenza polymerase PB2. Sci Rep. 2024; 14(1): 25441.https://doi.org/10.1038/s41598-024-69816-3
Sun Q, Fu Y, Wang W. Temperature effects on hydrophobic interactions: Implications for protein unfolding. Chem Phys. 2022; 559: 111550. https://doi.org/10.1016/j.chemphys.2022.111550
Bradford SYC, El Khoury L, Ge Y, Osato M, Mobley DL, Fischer M. Temperature artifacts in protein structures bias ligand binding predictions. Chem Sci. 2021; 12(34): 11275-11293 https://doi.org/ 10.1039/D1SC02751D
- Abstract Viewed: 424 times
- pdf Downloaded: 140 times